Clinical Summary
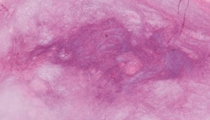
A 32-year-old man presents with a thigh mass of 6 months' duration. Physical exam reveals a large subcutaneous and deep firm, non-tender mass on his right lateral thigh. The lesion does not extend through the skin. CT-scan demonstrates a 6.3 cm solid mass in deep soft tissue of the right lateral thigh that focally extends into skeletal muscle but does not involve bone. The lesion is excised. Gross exam of the excision reveals a 6.2 x 5.7 x 5.1 cm well circumscribed, firm, tan homogeneous mass without hemorrhage or necrosis.
Master List
- Dermatofibrosarcoma protuberans, myxoid variant
- Fibromatosis
- Low-grade fibromyxoid sarcoma
- Ossifying fibromyxoid tumor
- Myxofibrosarcoma
- Sclerosing epithelioid fibrosarcoma
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2013, case 24, and is a low-grade fibromyxoid sarcoma.
Criteria for Diagnosis and Comments
Sections reveal a polymorphous mixed myxoid and fibroblastic spindle cell neoplasm. The fibroblastic component exhibits moderate cellularity with spindle cells arranged in irregular crisscrossing fascicles and storiform growth pattern. In some areas, the spindle cells have a wavy appearance. There is variable cellularity with hypocellular regions containing increased amount of collagenous stroma between spindle cells. The cells are bland appearing with light staining eosinophilic cytoplasm and oval to round slightly hyperchromatic nuclei. There is minimal nuclear pleomorphism with occasional small nucleoli and very minimal mitotic activity. Within the fibroblastic spindle cell component, there are regional areas of less cellular myxoid islands composed of bland spindle and stellate cells. A network of curvilinear vessels is present within the myxoid islands. No lipoblasts are present. The myxoid islands gradually transition into the more cellular fibroblastic component of the tumor. In some sections of the neoplasm, but not all, there are giant rosettes present composed of a central region of bright pink dense collagen bundles arranged somewhat haphazardly and surrounded by oval to round bland cells lacking nuclear atypia. Many of these cells have optically clear nuclei and rarely contain nuclear cytoplasmic pseudo-inclusions. The tumor does not exhibit necrosis, and most sections exhibit a fibrous outer wall with non-infiltrating margins. In view of scattered giant rosettes present in this neoplasm, one might favor classifying it as a hyalinizing spindle cell tumor with giant rosettes, which is considered a variant of low-grade fibromyxoid sarcoma.
Low-grade fibromyxoid sarcoma (LGFMS), also known as Evans tumor, is a soft tissue spindle cell neoplasm of probably myofibroblastic/fibroblastic origin having deceivingly bland histologic features but with metastatic potential. Clinically, this tumor is more common in males and can occur at any age but most occur in young and middle-aged adults. They usually arise in deep soft tissue involving skeletal muscle but some arise in superficial soft tissue with reports that superficial variants are more likely to be seen in children. Common sites include thigh (most common), chest wall, shoulder/axilla, pelvic region and neck. Unusual sites reported include mediastinum, mesentery, paravertebral, retroperitoneum, intracranial, lung and ovary. Clinically, they present as a slowly enlarging, painless, firm, variable-sized soft tissue mass of variable duration ranging from months to years.
Besides the histologic findings in this case, these tumors may exhibit areas with features of intermediate grade fibrosarcoma and necrosis. Mitotic activity is not a feature of this tumor. Mitotic counts are usually less than one 1 per 50 high power fields with one study recording an average of 1.6 mitoses in 50 high power fields. The classic giant rosette is described as a central region of collagen fibers arranged in a centripetal fashion from the center. The core of collagen is surrounded by oval to round cells having clear to pink cytoplasm, lacks nuclear atypia and may have intranuclear cytoplasmic pseudo-inclusions. In this case, there is a transition of the myxoid areas to the cellular areas, but in some cases the myxoid regions may abut suddenly with the fibroblastic areas without a transitioning region. In most tumors, the myxoid component does not predominate.
Immunohistochemical profiles have not been consistent but one marker, MUC4, has recently been shown to be a highly sensitive and specific immunohistochemical marker for LGFMS and sclerolsing epithelioid fibrosarcoma, and can help distinguish this tumor type from other histologic mimics. In addition, tumor spindle cells focally express smooth muscle actin, muscle specific actin, and diffusely vimentin suggesting myofibroblastic differentiation. Some tumors focally express EMA, CD68, CD34, AE1/AE3, and desmin but LGFMS does not express beta-catenin. In giant rosettes, surrounding round epithelioid cells of the rosette express vimentin and sometimes Leu-7 (CDg57), S100, PGP 9.5 and neuron specific enolase suggesting neural phenotype of these cells.
Cell of origin of these tumors is not definitive but immunohistochemical expression suggests myofibroblast. Electron microscopy has not been specific but suggest myofibroblast. One study found similarities with perineuroma, and in tumors containing giant rosettes, a Schwann cell origin has been proposed. Cytogenetics has shown the presence of a translocation, t(7;16)(q33;p11), in tumors with and without giant rosettes, resulting in FUS-CREB3L2 fusion. A similar translocation has also been reported in sclerosing epithelioid fibrosarcoma suggesting the possibility that it has a relationship to LGFMS. A combination of immunohistochemical studies, electron microscopy and cytogenetic studies support that hyalinizing spindle cell tumor with giant rosettes is a variant of LGFMS or it represents a histologic spectrum.
LGFMS is considered a low-grade sarcoma despite the usual benign-like histology including near-absence of mitotic activity and absence of significant nuclear atypia. The presence of focal intermediate or high-grade sarcoma/fibrosarcoma does not appear to worsen the short-term prognosis of these tumors. There are no studies reflecting long-term prognosis of LGFMS with focal intermediate or focal high-grade sarcoma/fibrosarcoma. Studies have not shown the significance related to prognosis when large areas of high-grade sarcoma/fibrosarcoma are present in these tumors. Prognosis appears to be better in pediatric patients than in adult patients with LGFMS. Initial prognostic studies occurred on patients who seemed to have a high rate of local recurrence and metastases. However, these patients had not been treated as a sarcoma initially until there was a recurrence or metastasis. Later studies are on patients predominantly diagnosed initially as a sarcoma, receiving more aggressive surgery. Recurrence rate in these patients treated with aggressive surgery is approximately 7%, metastatic rate is about 4%, and death related to tumor is approximately 1-2%. Metastatic disease has been reported at the time of diagnosis, and metastases have occurred late in the course of the neoplasm up to four and one-half decades after initial diagnosis. As a result, patients with these tumors require follow-up for the remainder of their life. Wide surgical excision is the current treatment of choice when diagnosed.
When considering LGFMS as a diagnosis, several entities, both benign and malignant must be considered in the differential diagnosis. Many tumors containing myxoid foci need to be considered including dermatofibrosarcoma protuberans, myxofibrosarcoma, desmoid fibromatosis, aggressive angiomyxoma, myxoid liposarcoma, and myxoid neurofibroma. Other tumors with similar cytogenetic findings such as sclerosing epithelioid fibrosarcoma should also be considered.
Dermatofibrosarcoma protuberans (DFSP), myxoid variant, may have myxoid regions that mimic LGFMS, but DFSP regularly expresses CD34 while LGFMS may focally express CD34. DFSP exhibits a storiform spindle cell pattern while LGFMS does not. DFSP is superficial while some cases of LGFMS may be superficial, particularly in pediatric patients; most cases of LGFMS are in deep soft tissue.
Desmoid fibromatosis may be difficult to distinguish from LGFMS histologically as both are composed of slender, bland spindle cells that express vimentin, smooth muscle actin and muscle specific actin pointing towards myofibroblastic differentiation. Prominent hyalinization and glassy keloid-like collagen may be present in fibromatosis. Fibromatosis also regularly expresses beta-catenin, as part of the APC gene mutation, while LGFMS does not.
Myxofibrosarcoma usually arises clinically in the superficial soft tissue of extremities of elderly patients instead of deep soft tissue of younger patients seen in LGFMS. Myxofibrosarcoma exhibits a greater degree of nuclear atypia than LGFMS, and it is predominantly myxoid lacking the fibrous/fibroblastic regions of LGFMS. Immunohistochemically, myxofibrosarcoma strongly expresses vimentin and focally expresses actins similar to LGFMS. No tumor specific cytogenetic finding has been determined for myxofibrosarcoma, however, cytogentic studies have demonstrated very complex triploid and tetraploid karyotypes in these tumors, but they are FUS negative by FISH.
Ossifying fibromyxoid tumor (OFT) is often thought to be in the differential diagnosis of LGFMS and has morphologic nuclear overlap. It usually is in a subcutaneous or intramuscular location in an adult with a partial rim of bone in most cases. Some cases of LGFMS have bone surrounding the tumor as well. OFT can also have myxoid foci and prominent vessels, like LGFMS. The similar bland ovoid nuclei of OFT usually have distinctive cytoplasmic borders unlike LGFMS and would be FUS negative and sometimes S-100 protein positive.
With similar cytogenetic and immunophenotypic findings, sclerosing epithelioid fibrosarcoma should be considered in the differential diagnosis since these tumors may also have myxoid patterns present. Most of these tumors arise in deep soft tissue of extremities and may involve bone while LGFMS does not. In non-myxoid zones, these tumors are hypocellular and are composed of epithelioid cells having clear to faint eosinophilic cytoplasm. Cells are separated by dense collagenous stroma, which sometimes diminishes or appears to eradicate the epithelioid tumor cells. These tumors generally do not express actins or cytokeratins but may express MUC4 and EMA, sometimes resulting in confusion that the tumor is a carcinoma.
Other tumors that might be considered in the differential include aggressive angiomyxoma, myxoid neurofibroma, myxoid liposarcoma and perineurioma all lacking cytogenetic evidence of a FUS translocation. Aggressive angiomyxoma, usually located in the perineum, lacks a fibrous/fibroblastic pattern and is predominantly myxoid containing large vessels not seen in LGFMS. It also regularly expresses desmin. In myxoid neurofibroma, cells exhibit more prominent nuclear waviness than in LGFMS, and cells strongly express S100. In perineurioma, myxoid regions are infrequent and cells diffusely express EMA. With a capillary network present in both myxoid liposarcomas and LGFMS, both tumors may be similar. No lipoblasts are present in LGFMS, and the capillary network in LGFMS is composed of ones that are curvilinear that is atypical for myxoid liposarcoma.
Supplementary Questions
- Low-grade fibromyxoid sarcoma exhibits no nuclear atypia, brisk mitotic activity, and consistently expresses EMA and beta catenin.
- True
- False
- Cytogenetic studies have suggested that sclerosing epithelioid fibrosarcoma may be related to low-grade fibromyxoid sarcoma
- True
- False
- Low-grade fibromyxoid sarcoma in pediatric patients are more likely to be in superficial soft tissue as compared with deep soft tissue seen in adult tumors.
- True
- False
References
- Billings SD, Giblen G, Fanburg-Smith JC. Superficial low-grade fibromyxoid sarcoma (Evans tumor): a clinicopathologic analysis of 19 cases with unique observation in the pediatric population. Am J Surg Pathol. 2005;29:204-210.
- Doyle LA, Möller E, Dal Cin P, et al. MUC4 is a highly sensitive and specific marker for low-grade fibromyxoid sarcoma. Am J Surg Pathol. 2011;35(5):733-41.
- Evans HL. Low-grade fibromyxoid sarcoma. A report of 12 cases. Am J Surg Pathol. 1993;17:595-600.
- Evans HL. Low-grade fibromyxoid sarcoma. A report of two metastasizing neoplasms having deceptive benign appearance. Am J Clin Pathol. 1987;88:615-619.
- Folk GS, Williams SB, Foss RB, Fanburg-Smith JC. Oral and maxillofacial sclerosing epithelioid fibrosarcoma: Report of five cases. Head and Neck Pathol. 2007;1:13-20.
- Folpe AL, Lane KL, Pauli G, et al. Low-grade fibromyxoid sarcoma and hyalizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high grade areas. Am J Surg Pathol. 2000; 24:1353-1360.
- Lane KL, Shannon RJ, Weiss SW. Hyalinizing spindle cell tumor with giant rosettes: a distinctive tumor closely resembling low-grade fibromyxoid sarcoma. Am J Surg Pathol.1997;21:1481-1488.
- Tun K, Ozen O, Kaptanoglu E, et al. Primary intracranial low-grade fibromyxoid sarcoma (Evans tumor). J Clin Neurosci. 2008;15:1298-1301.
- Vernon SE, Bejarano. Low-grade fibromyxoid sarcoma, a brief review. Arch Pathol Lab Med. 2006; 130:1358-1360.
- Weiss SW, Goldblum JR. Enzinger and Weiss's Soft Tissue Tumors, Fifth Edition. China, Mosby-Elsevier, 2008.
Author
2013
Byron E. Crawford, MD
Surgical Pathology Committee
Tulane University School of Medicine
New Orleans, LA
Answer Key
- False (b).
- True (a).
- True (a).