Clinical Summary
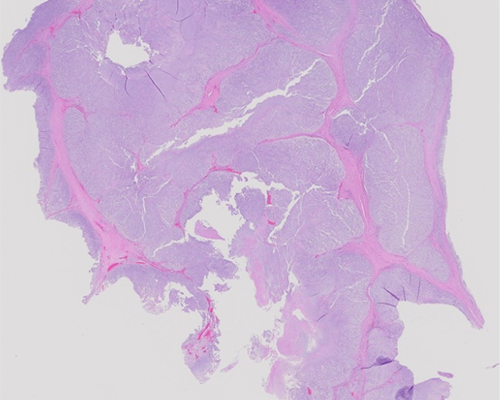
A 57-year-old man presents with a dull lower back pain for the last six months, which worsens while sitting. Clinical workup and imaging reveals an 8.0 cm mass in the sacrum area. The patient undergoes an excision of the mass. Grossly, the mass is soft and lobulated with a gelatinous cut surface. By immunohistochemistry, the tumor cells stain positive for S100 protein and pan-keratin.
Master List of Diagnoses
- Chondrosarcoma
- Chordoma
- Metastatic carcinoma
- Myxopapillary ependymoma
- Parachordoma/mixed tumor/myoepithelioma of soft tissue
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2016, case 40, and is a chordoma.
Criteria for Diagnosis and Comments
Sections show lobules of tumor cells in a background of myxoid matrix separated by fibrous septa. The tumor cells are arranged in cords and have small nuclei and abundant cytoplasm. Some of the tumor cells have bubbly cytoplasm, referred to physaliferous cells. These histologic features are diagnostic of chordoma.
Chordomas are rare tumors of bone with a reported incidence of approximately 1:1,000,000. Chordomas are the only malignant tumors arising from the notochord elements and occur in the midline of the body. These tumors affect males twice as often as females and tend to occur at either end of the spinal column. When they occur in the vertebrae, the majority involve the cervical vertebrae and rarely thoracic vertebrae. Rarely these can be seen in children, most often in the skull base. They are thought to arise from notochord remnants but more recently it is suggested that they arise from benign notochord cell “tumors” with inherent neoplastic potential. Chordomas are slow-growing, gelatinous, extradural tumors with areas of hemorrhage and necrosis, often large in size. They cause late-onset compressive neurological symptoms. Clonal chromosomal abnormalities including loss of chromosomes 3, 4, 10, and 13, and various deleted segments, have been reported. Microsatellite instability resulting from DNA mismatch repair genes has also been reported.
Chordomas show well-characterized imaging findings including a solitary, central, lytic, destructive lesions of the axial skeleton associated with a soft tissue mass. They often have intratumoral calcifications. On microscopic examination, lobulation and cording of cells are key to diagnosing chordomas. The tumor cells are arranged in sheets, cords, or singly within a myxoid or mucoid stroma, which stains positive with alcian blue and mucicarmine. The pathognomonic physaliferous cells have a small, dark round to ovoid nucleus and abundant multivacuolated cytoplasm, which rarely can be more solid and eosinophilic with mild to moderate nuclear pleomorphism. Necrosis is infrequent, and mitoses are usually rare. Areas of calcification, hemorrhage, and hemosiderin deposition can often be identified. Cases with prominent epithelioid or spindle cell sarcomatoid components ("dedifferentiated" chordoma or sarcomatoid chordoma) with bizarre cytological atypia have also been reported in less than 5% of cases. The tumor cells stain positive with S100 protein, keratins, and brachyury (marker of notochordal origin). The chordomas of the base of the skull can show prominent chondroid differentiation; these chondroid chordomas are associated with much better prognosis than conventional chordomas.
The histologic differential diagnosis includes myxopapillary ependymoma, metastatic carcinoma, chondrosarcoma, and parachordomas/mixed (myoepithelial) tumors. The cells of chordomas may appear epitheliod and bear resemblance to metastatic carcinoma. However, it is unusual to see a distinctive lobulated growth pattern and fibrous septa in metastatic carcinoma. Both these tumors stain positive with keratins, but chordomas additionally stain positive with S100 protein.
Chondrosarcomas tend to be lobulated, but they do not show fibrous septa and are negative for epithelial markers. Myxopapillary ependymomas also stain negative with epithelial markers.
Parachordoma/mixed tumor/myoepithelioma of soft tissue may resemble chordomas and extraskeletal myxoid chondrosarcoma, however these tumors show a ductal component in 15% to 20% of cases with relative proportions of chondromyxoid/myxoid tissue. They usually show well-circumscribed lobules of large, round cells with eosinophilic cytoplasm, focally resembling physaliferous cells in myxoid to densely hyaline matrix. They show a blend of vacuolated cells with spindled cells and small glomoid cells. Chordomas express CK1/10 and CK19, both of which are absent in parachordomas. In addition, myxoid chondrosarcomas and mixed tumors occur in soft tissues and not in the midline.
Chordomas have a poor response to radiotherapy or chemotherapy but are amenable to surgical excision. These tumors can locally recur, with a low potential for metastasis. Survival is determined by the success or failure of local control; therefore, early diagnosis is crucial. Transformation of chordoma into a high-grade spindle cell sarcoma (dedifferentiation) has been described with and without radiation therapy.
Supplementary Questions
- Which of the following statements accurately describes the most common locations for chordomas?
- Most chordomas arise in the extremities.
- Most chordomas arise in the head and neck region.
- Most chordomas arise in the midline, at either end of the spinal cord.
- Most chordomas arise within the peritoneum.
- Which of the following immunoprofiles does a chordoma most commonly exhibit?
- Brachyury negative, S100 protein negative, pan-keratin negative.
- Brachyury negative, S100 protein negative, pan-keratin positive.
- Brachyury negative, S100 protein positive, pan-keratin negative.
- Brachyury positive, S100 protein positive, pan-keratin positive.
- Which of the following statements describes chordomas accurately?
- Chordomas are rare tumors of the bone with a predilection for males.
- Chordomas never metastasize.
- Chordomas occur in the epiphysis of the long bones.
- Patients presenting with chordomas experience no symptoms.
References
- Fletcher, CDM, Bridge, JA, Hogendoorn, P, Mertens F. World Health Organization Classification of Tumours: Pathology and Genetics of Tumors of Soft Tissue and Bone. Lyon, FR: IARC Press; 2013.
- Inwards CY, Oliveira AM. Tumors of the osteoarticular system. In Fletcher CD, ed. Diagnostic Histopathology of Tumors, 4th edition. Philadelphia, PA: Elsevier; 2013:1871-1932.
- Meis JM, Raymond AK, Evans HL, Charles RE, Giraldo AA. "Dedifferentiated" chordoma. a clinico-pathologic and immunohistochemical study of three cases. Am J Surg Pathol. 1987;11:516-525.
Author
2016
Kirtee Raparia, MD
Surgical Pathology Committee
Northwestern University
Chicago, IL
Answer Key
- Most chordomas arise in the midline, at either end of the spinal cord. (c)
- Brachyury positive, S100 protein positive, pan-keratin positive (d)
- Chordomas are rare tumors of the bone with a predilection for males. (a)