Clinical Summary
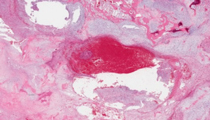
A 65-year-old man presents with a painful left pelvic mass of four months duration. The pain is intermittent, is noted at rest and becomes worst at night. Radiographs reveal an 8.4 cm intramedullary lucent lesion of the left pelvic bone that contains irregular punctate opacities. The lesion extends through the cortex that demonstrates prominent adjacent periosteal reaction, and involves soft tissue. There is no history of previous radiation to this area. The lesion is resected and represented by the enclosed slide.
Master List
- Chondroblastic osteosarcoma
- Chondroblastoma
- Chondrosarcoma, grade 2
- Dedifferentiated chondrosarcoma
- Enchondroma
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2013, case 04, and is a chondrosarcoma, grade 2.
Criteria for Diagnosis and Comments
Sections show lobules of hypercellular hyaline cartilage permeating around and surrounding bony trabeculae and in some sections destruction of the cortex with extension into the periosteum. The cells are present within lacunae and exhibit moderate nuclear pleomorphism with occasional prominent nucleoli and binucleation. Some nuclei contain open chromatin patterns. Multiple foci of myxoid patterns are present. The clinical and histologic findings are diagnostic of a chondrosarcoma, grade 2.
Chondrosarcoma is a malignant tumor with pure hyaline cartilage differentiation. They may be subclassified into primary (conventional) types or secondary types, which arise from enchondromas or osteochondromas. Primary chondrosarcomas are the third most common primary malignancy of bone accounting for approximately 20% of malignant bone tumors. Chondrosarcomas are predominately a disease of older individuals with peak incidence in the 5th to 7th decade. The most common presenting symptoms are pain and swelling, and are usually of long duration (months to years).
The most common sites are the bones of the pelvis (ilium most frequent), proximal and distal femur, proximal humerus and ribs. In the long bones, they are generally metaphyseal or diaphyseal lesions and in the ilium they usually occur near the acetabulum. Chondrosarcomas rarely occur in the bones distal to the wrist and ankles or in the skull and jaw. As with all bone neoplasms, radiographic findings are very important. Radiographic findings of chondrosarcomas include radiolucent lesions containing calcifications reflected as ring-like or punctate opacities. There may be cortical deep endosteal erosions or complete cortical disruption with tumor extending into soft tissue. Any chondroid tumor with cortical erosion, cortical thinning or thickening, expansion of bone cortex, endosteal scalloping (reabsorption of endosteal cortex), or production of reactive periosteal new bone formation on radiographs should suggest an aggressive tumor.
Macroscopically, chondrosarcomas are lobulated light blue or white masses corresponding to hyaline cartilage production. There may be foci of myxoid change. Chalky white material represents calcium deposits. Histologic examination reveals irregular lobules of abundant blue-gray cartilage matrix with overall hypercellularity, although cellularity may vary from field to field. The lobules of cartilage permeate the medullary space and surround bony trabeculae. Chondrocytes within lacunae exhibit variable atypia with variation in size and shape, and may have open-chromatin patterns. Binucleation is frequently seen and must be distinguished from multiple cells within a single lacuna. Chondrosarcomas express S-100, estrogen receptors and SOX9, a regulator of chondrogenesis. BCL2 is expressed in approximately fifty percent of chondrosarcomas.
Chondrosarcomas are divided into three grades. Several studies have shown the prognostic significance of histologic grading. About half of all chondrosarcomas are grade 1, 40% are grade 2 and the remainder are grade 3. Grading is based on cellularity, nuclear size and nuclear hyperchromasia. Grade 1 tumors are moderately cellular with hyperchromatic slightly enlarged nuclei. Grade 2 tumors are more cellular with greater degree of nuclear atypia and hyperchromasia. Tumors with myxoid features are classified as grade 2 even with low cellularity. Grade 3 tumors are very cellular with pleomorphic nuclei and frequently identified mitoses. However, sheets of spindle cells are not present.
Chondrosarcomas must be differentiated from multiple benign and malignant tumors that produce a hyaline cartilage matrix such as enchondromas and chondroblastic osteosarcomas. Enchondromas are benign intramedullary hyaline cartilage tumors that can be difficult at times to differentiate from a low-grade chondrosarcoma based on histology alone. In contrast to chondrosarcomas, enchondromas are usually asymptomatic, and commonly occur in the hands and feet, and are rare in the pelvis and ribs. Enchondromas are typically discovered incidentally on radiographs. Radiographically, they are expansile lucent nodules that may have characteristic punctate, stippled or ring mineralization. Histologically, they are generally lobulated hypocellular tumors with abundant mature cartilage although those that occur in the small bones of hand and feet may be hypercellular. Cells have small uniform, dark nuclei, and granular or vacuolated cytoplasm. Binucleated chondrocytes and nuclei with open chromatin patterns may be found but are not prominent. They grow as lobules between bone marrow elements and bone trabeculae without entrapping trabeculae. No myxoid foci are seen. The presence of pain with a more proximal location towards the axial or appendicular skeleton in a tumor with histologic features suggestive of an enchondroma should raise suspicion of a low grade malignancy unless a pathologic fracture has occurred in the tumor.
Chondroblastic osteosarcoma is in the differential diagnosis due to its predominant chondroid component. The chondroid component can be very extensive and variably represented by high or intermediate grade patterns. By definition, at least a small focus of tumor osteoid must be present to qualify as a chondroblastic osteosarcoma. Clinical setting and radiographic findings are helpful in distinguishing chondroblastic osteosarcoma from chondrosarcoma. Osteosarcomas of all types would be unusual in an elderly patient such as this one as most are seen in second and third decades of life. Osteosarcoma arising in older patients outside of this age group should suggest preexisting conditions including Paget’s disease of bone (multi-focal osteosarcoma), Li-Fraumeni syndrome, and past radiation to the affected bone. It has been reported that osteosarcoma including chondroblastic osteosarcoma expresses ezrin (a cytoskeletal protein) while chondrosarcomas are negative. Ezrin is a potential immunohistochemical marker that may be used to help differentiate the two neoplasms that may have similar histology in biopsies. Chondroblastic osteosarcoma may be seen in craniofacial bones in the third and fourth decades of life.
Dedifferentiated chondrosarcomas are bimorphic malignant neoplasms in which chondrosarcoma is adjacent to a malignant spindle cell neoplasm. The two morphologic patterns are sharply demarcated and the cartilage component does not blend into the spindle cell component as in osteoblastic osteosarcoma. The chondrosarcoma typically has features of grade 1 tumors. The spindle cell sarcomatous component is hypercellular with histomorphology of a malignant fibrous histiocytoma, fibrosarcoma, osteosarcoma or rhabdomyosarcoma. On gross examination, the spindle cell sarcoma may be recognized as a tan fleshy mass generally in an extraosseous location with typical findings of the intramedullary chondrosarcoma. The prognosis of dedifferentiated chondrosarcoma is dismal, underscoring the importance of sampling any tan to white fleshy areas at gross dissection.
Chondroblastomas commonly occur in the epiphyses of skeletally immature individuals with a male predominance. They can be painful and may recur with secondary aneurysmal bone cyst formation. Radiographically, these tumors produce a radiolucent or lytic epiphyseal defect with a rim of sclerotic bone. Chondroblastomas are composed of monomophic mononuclear cells, and occasional giant cells within a scant immature hyaline type chondroid matrix. The mononuclear cells represent embryonic chondroblasts that contain lobulated indented nuclei with some mitotic activity, but without atypical forms.
The presence of the mononuclear cells and multinucleated giant cells may mimic a giant cell tumor histologically. Calcification of the matrix may occur, giving rise to the classic "chicken-wire" appearance in which linear calcifications surround individual mononuclear cells. Chondroblastoma expresses S-100 and SOX9.
Supplementary Questions:
- Osteosarcoma arising in a 66-year-old man may be associated with Paget's disease of bone or Li-Fraumeni syndrome.
- True
- False
- Chondroblastoma may produce "chicken wire" linear calcifications around mononuclear tumor cells.
- True
- False
- Primary chondrosarcomas are the most common primary malignancy of bone.
- True
- False
References
- Dorfman HD, Czerniak B. Bone Tumors. St. Louis: Mosby, 1998.
- Fletcher DM, Unni KK, Mertens F. WHO Classification of Tumors: Pathology and Genetics of Tumors of the Soft Tissue and Bone. Lyon: IARC Press, 2002.
- Rosai, J. Rosai and Ackerman's Surgical Pathology. Volume 2, 10th edition. China: Mosby Elsevier, 2011.
- Unni KK, Inwards CY, Bridge JA, Kindblow L, Wold L. AFIP Atlas of Tumor Pathology: Tumors of the Bone and Joints, 4th Series. Silver Spring: ARP Press, 2005.
Authors
2012
Jonathan Stone, MD
Department of Pathology and laboratory Medicine
Tulane University School of Medicine
Byron Crawford, MD
Surgical Pathology Committee
Tulane University School of Medicine
New Orleans, LA
Answer Key
- True (a).
- True (a).
- False (b).