- Home
- Member Resources
- Pathology Case Challenge
- Left Arm
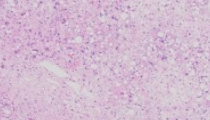
A 72-year-old woman presents with a 7.0 x 3.5 x 3.0 cm mass in the deep soft tissues of the upper extremity. She has a prior history of malignancy but cannot recall what type it was. There is no additional clinical information available for the patient.
Master List of Diagnoses
- Dedifferentiated liposarcoma
- Metastatic gastric carcinoma (signet ring cell type)
- Metastatic renal cell carcinoma
- Myxofibrosarcoma, high-grade
- Myxoid/round cell liposarcoma
- Pleomorphic lipoma
- Pleomorphic liposarcoma
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2016, case 07, and is a pleomorphic liposarcoma.
Criteria for Diagnosis and Comments
The section provided shows a soft tissue neoplasm characterized by numerous large pleomorphic tumor cells. There are areas of lipoblasts as well as areas with a looser, myxoid type of stroma. Lipoblasts are identified by the presence of large fat vacuoles which tend to displace the cell nucleus to the periphery. The lipoblasts contain a single fat vacuole or, less commonly, can be multi-vacuolated. In areas without obvious lipoblasts there are numerous large pleomorphic tumor cells. Numerous multinucleate cells are identified, often with “wreath-like” arrangements. In addition, mitotic activity is increased, and atypical mitotic figures are easily identified.
Pleomorphic liposarcoma (PLS) represents the rarest of the major subtypes of liposarcoma, probably accounting for about 5% of all liposarcomas. This is a high-grade, aggressive neoplasm with a propensity for multiple recurrences and metastases. PLS is largely a disease of older individuals but rare cases in younger patients, even adolescents, have been recorded. As true of many other varieties of sarcoma, the most common site of the tumor is the deep soft tissues of the proximal extremity. However, this tumor has been reported in a number of unusual sites as well, including the mediastinum, head and neck region, and pericardium.
Unlike the other major subtypes of liposarcoma, PLS does not characteristically show a reproducible cytogenetic or molecular abnormality. Instead, PLS is characterized by multiple chromosome deletions, translocations, and additions. Lack of a recurrent abnormality makes development of a “targetable” therapy for PLS problematic. Treatment is largely surgical, and prognosis tends to be poor. Patients usually develop metastatic disease, commonly in the lungs.
The differential diagnosis of PLS is broad and includes a number of other mesenchymal neoplasms in addition to carcinomas with extensive clear cell change. Pleomorphic lipoma is a benign entity that overlaps extensively with the spindle cell variant of lipoma. Pleomorphic/spindle cell lipoma tends to occur in the head, neck, and trunk of older men (mean age 55) and tends to manifest as clinically a circumscribed, subcutaneous mass. This variant of lipoma rarely exceeds 5.0 cm in size. Like PLS, the pleomorphic subtype of lipoma can show nuclear atypia and multinucleation, often in a “floret-like” pattern. Unlike PLS, pleomorphic/spindle cell lipoma will usually show diffuse and strong immunohistochemical staining for CD34. In addition, pleomorphic/spindle cell lipoma is usually a circumscribed lesion whereas PLS tends to be an infiltrative or poorly delineated tumor. Also, pleomorphic/spindle cell lipomas often show monosomy or partial loss of chromosome 13.
Dedifferentiated liposarcoma (DDLS) represents an additional diagnostic consideration in this case. DDLS tends to occur in the setting of a pre-existing well-differentiated liposarcoma but can occasionally present de novo. DDLS most commonly occurs in the retroperitoneum. Histologically, DDLS will most often resemble an undifferentiated pleomorphic sarcoma or a myxofibrosarcoma, entities unlikely to be confused with PLS. In some instances, however, DDLS can show focal areas of “homologous” lipoblastic differentiation which can assume a “pleomorphic liposarcoma-like” appearance. In this instance, demonstration of MDM2 and CDK4 amplification, either by immunohistochemical analysis or by FISH, would be helpful in making a distinction between true PLS and DDLS with PLS-like morphology.
A high-grade myxoid liposarcoma (MLS) with an extensive “round cell” component may also be confused with PLS. A subset of classic MLS will display a variant pattern with a predominance of “round cells”. The round cell element is a histologically more primitive cell, arranged in a more cellular configuration, sometimes with lipoblasts, but often without obvious adipocytic differentiation. If 5% or more of the total tumor is comprised of the "round cell" pattern, the tumor tends to be associated with a more aggressive behavior than conventional MLS. The round cell component has a higher nuclear to cytoplasmic ratio and can be confused with other “small round blue cell” tumors such as lymphoma or Ewing sarcoma. Fortunately, the myxoid/round cell subtype of liposarcoma contains a distinctive cytogenetic signature which helps differentiate this neoplasm from other histologic mimics. The myxoid/round cell variant of liposarcoma is characterized cytogenetically by a balanced translocation, most commonly between chromosomes 12 and 16. The t(12;16)(q13;p11) translocation leads to fusion of the DDIT3 gene locus (also known as CHOP) to a partner gene, most often FUS. EWSR1 represents an alternative partner gene for DDIT3, identified in about 5% of cases.
PLS is also likely to be confused with high-grade myxofibrosarcoma. Both of these tumors are sarcomas that contain an abundance of extracellular myxoid material. Myxofibrosarcoma will often have multinucleate giant cells as well as vacuolated malignant cells called “pseudolipoblasts”. In contrast to myxofibrosarcoma, PLS will show large foci or aggregates of pleomorphic lipoblasts instead of single, isolated, lipoblast-like cells. Both PLS and myxoid liposarcoma have complex cytogenetic profiles and, to date, there is not a reliable molecular or cytogenetic feature to help distinguish them. Adequate sampling to demonstrate all histologic features is the best way to differentiate these two neoplasms.
And lastly, the presence of numerous vacuolated cells may raise the possibility of a metastatic carcinoma, particularly given the history of a previous (unknown type) malignancy in this patient. Metastatic renal cell, adrenal carcinoma, and metastatic signet ring cell carcinomas could potentially be confused with PLS. Adrenal and renal cell carcinomas are often characterized by abundant clear cytoplasm, but individual cells would rarely should a “lipoblastic” appearance with well-defined cytoplasmic vacuoles. Signet ring variants of adenocarcinoma arise throughout the gastrointestinal tract and are particularly common in the stomach. Both of these neoplasms can occasionally present clinically as a metastasis to an unusual site such as soft tissue prior to clinical detection of the primary tumor. To further complicate this issue, there are rare reports of a so-called “epithelioid” variant of PLS that will occasionally stain focally positive for EMA. In this instance, immunohistochemical staining for keratin can be useful as an epithelioid variant of PLS should not be immunoreactive. PLS will also show focal S100 immunoreactivity within the lipoblastic foci; a feature absent in carcinomas with clear cell cytoplasm.
Supplementary Questions:
- Which of the following ancillary studies is most likely to help distinguish pleomorphic liposarcoma from pleomorphic lipoma?
- FISH for EWSR1 translocation
- FISH for FUS translocation
- Immunohistochemical staining for CD34
- Immunohistochemical staining for MDM2 and CDK4
- Which of the following molecular tests is most helpful in distinguishing pleomorphic liposarcoma from de-differentiated liposarcoma?
- FISH for amplification of MDM2
- FISH for translocation of MDM2
- Immunohistochemical staining for CD34
- Immunohistochemical staining for S100
- Which is the best way to differentiate pleomorphic liposarcoma from high-grade myxofibrosarcoma?
- FISH for a balanced translocation
- Histologic sampling to identify areas with lipoblasts
- Histologic sampling to identify myxoid matrix material
- Karyotype analysis of tumor cells
References
- Downes KA, Goldblum JR, Montgomery EA, Fisher C. Pleomorphic liposarcoma: a clinicopathologic analysis of 19 cases. Mod Pathol.2001;14:179-184.
- Gebhard S, Coindre JM, Michels JJ, et al. Pleomorphic liposarcoma: clinicopathologic, immunohistochemical, and follow-up analysis of 63 cases: a study from the French Federation of Cancer Centers Sarcoma Group. Am J Surg Pathol.2002;26:601-616.
- Ghadimi MP, Liu P, Peng T, et al. Pleomorphic liposarcoma: clinical observations and molecular variables. Cancer. 2011;117:5359-5369.
- Idbaih A, Coindre JM, Derre J, et al. Myxoid malignant fibrous histiocytoma and pleomorphic liposarcoma share very similar genomic imbalances. Lab Invest. 2005;85:176-181.
- Kilpatrick SE, Doyon J, Choong PF, Sim FH, Nascimento AG. The clinicopathologic spectrum of myxoid and round cell liposarcoma. A study of 95 cases. Cancer.1996;77:1450-1458.
- Mariño-Enriquez A, Fletcher CD, Dal Cin P, Hornick JL. Dedifferentiated liposarcoma with “homologous” lipoblastic (pleomorphic liposarcoma-like) differentiation: Clinicopathologic and molecular analysis of a series suggesting revised diagnostic criteria. Am J Surg Pathol. 2010;34:1122-1131.
- Miettinen M, Enzinger FM. Epithelioid variant of pleomorphic liposarcoma: a study of 12 cases of a distinctive variant of high-grade liposarcoma. Mod Pathol.1999;12:722-728.
Author
2016
Leslie G Dodd, MD
Surgical Pathology Committee
University of North Carolina
Chapel Hill, NC
Answer Key
- Immunohistochemical staining for CD34 (c)
- FISH for amplification of MDM2 (a)
- Histologic sampling to identify areas with lipoblasts (b)