Clinical Summary
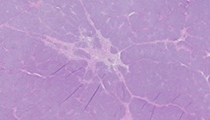
A 9-month-old boy presents with a painless abdominal mass. Imaging reveals a left renal mass and a left total nephrectomy is performed subsequently. Gross examination reveals a 6.5 cm lower pole tumor with a lobulated, bulging, rubbery tan cut surface.
Master List
- Clear cell sarcoma
- Lymphoma/leukemia
- Metanephric adenoma
- Nephroblastoma (Wilms tumor)
- Primitive neuroectodermal tumor
- Rhabdoid tumor
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2014, case 20 and is a nephroblastoma (Wilms tumor).
Criteria for Diagnosis and Comments
This kidney tumor is composed of nests of small, closely-packed round to polygonal cells consisting of small and overlapping nuclei showing evenly distributed chromatin and small nucleoli and scant cytoplasm. Numerous mitotic figures are present. There are no anaplastic foci. This case differs from most nephroblastomas (characteristically triphasic with blastemic, epithelial and stromal elements) in that it is composed mostly of blastema.
Nephroblastoma is a malignant embryonal neoplasm derived from nephrogenic blastemal cells. It is the most common genitourinary cancer in children, comprising approximately 85% of all pediatric renal neoplasms. Nephroblastoma occurs with equal frequency in both kidneys. The clinical presentation is usually a painless abdominal mass. The gross appearance is typically a solitary, well-circumscribed mass with a fibrous pseudocapsule. The cut surface varies from uniform, pale gray or tan with soft consistency, to firm and whorled, particularly if a large fraction of tumor is composed of stromal components. Microscopically, most nephroblastomas are triphasic, containing undifferentiated blastemal cells and differentiated epithelial and stromal elements. The blastemal cells occur in several different patterns: 1) diffuse, 2) nodular, 3) serpentine, and 4) basaloid. The nodular and serpentine blastemal patterns are the most frequently encountered and are seen as the predominant patterns in this case. An epithelial component of differentiation is usually present, including primitive rosette-like structures, tubular or papillary elements. The stromal component is often comprised of spindle cells in a myxoid background. Heterologous stromal components consisting of skeletal muscle, smooth muscle, cartilage, bone, and adipose tissue can also be seen.
It is important to make the distinction between tumors with favorable histology versus unfavorable histology since the former is highly responsive to chemotherapy. Unfavorable histology is defined as the presence of nuclear anaplasia or rarely, the development of high-grade carcinoma or sarcoma within a nephroblastoma. Nuclear anaplasia is present when there is nuclear hyperchromasia, enlargement (at least 3x the size of nonneoplastic nuclei involving all diameters), and multipolar mitotic figures. These features are absent in this case.
In nephroblastoma, deletion of chromosome 11p13 involving the WT1 gene was first recognized in patients with WAGR syndrome (Wilms tumor, aniridia, genitourinary malformation, and mental retardation). Germline point mutations in this gene are recognized in Denys–Drash syndrome (Wilms tumor, pseudohermaphroditism, and mesangial sclerosis).
The differential diagnosis of nephroblastoma includes but is not limited to: metanephric adenoma, clear cell sarcoma, rhabdoid tumor, primitive neuroectodermal tumor, and lymphoma/leukemia. Morphologic features are usually sufficient for accurate classification of nephroblastomas; however, immunohistochemistry may be of use when it exhibits overlapping morphology with other entities in the usual differential diagnoses. The blastema cells usually express vimentin, and may show focal positive reactivity with desmin and cytokeratin. WT-1 is confined to the nucleus with a higher concentration of positivity in the blastema cells and lower to no expression in areas of stromal and terminal epithelial differentiation.
Metanephric adenoma is a benign tumor composed exclusively of epithelial nephroblastic cells. There is a female predominance and most patients are in their fifth or sixth decade at time of diagnosis. However, the reported age range is 15 months to 83 years. Most often the tumor is found incidentally, but can present as a mass. There has been an association with polycythemia in about 12% of cases reported. Grossly, the tumor is unencapsulated, tan to yellow, soft or firm with frequent calcifications. Microscopically, metanephric adenoma is composed of small, uniform, epithelial cells that form small acini separated by acellular stroma. The commonly overlapping tumor cells have scant pale-staining cytoplasm, dark nuclei, and inconspicuous nucleoli. Mitotic figures are rare. Metanephric adenoma can mimic epithelial-predominant nephroblastoma and the solid variant of papillary renal cell carcinoma, especially in adult patients. Immunohistochemical and cytogenetic studies are useful to distinguish between these entities. Table 1 outlines the immunoreactivity for these entities.
| CK7 | CD57 | AMACR | WT-1 | |
|---|---|---|---|---|
| Metanephric adenoma | - | + | - | + |
| Nephroblastoma | - | - | - | + |
| Papillary RCC, solid | + | - | + | - |
AMACR – alpha-methylacyl-CoA racemase; WT-1 – Wilms Tumor-1 protein; RCC – renal cell carcinoma
Clear cell sarcoma of the kidney (CCSK) comprises about 3% of all pediatric renal neoplasms and the mean age at diagnosis is 3 years. Grossly, CCSK is a large unicentric mass that is well-circumscribed and sharply demarcated from adjacent kidney. Microscopically, CCSK consists of undifferentiated cells with abundant extracellular matrix that are separated into cords and nests by fine fibrovascular septa. One helpful feature of CCSK is the microscopic appearance of the kidney-tumor interface demonstrating highly infiltrative tumor cells invading the normal renal parenchyma, in contrast to other encapsulated malignant pediatric renal neoplasms. There are several variant patterns of CCSK including: 1) myxoid, 2) sclerosing, 3) cellular, 4) epithelioid, 5) palisading, 6) storiform, and 7) spindle cell. The cellular and epithelioid patterns of CCSK may simulate the blastemal and tubule components of nephroblastoma, respectively. Careful attention to the presence of regularly spaced fibrovascular septa present in CCSK is helpful in making the distinction. Immunohistochemically, the cord cells of CCSK usually demonstrate positive staining for vimentin. S100, desmin, CD99, cytokeratin, and epithelial membrane antigen are uniformly negative.
Rhabdoid tumor of the kidney (RTK) is aggressive and lethal, comprising about 2–3% of malignant renal tumors in children. The median age of diagnosis is 1 year. Grossly, the tumor is usually unicentric and unilateral, bulging, soft and pale, sometimes with hemorrhage or necrosis, and has an ill-defined kidney-tumor interface. Microscopically, this tumor is composed of monotonous sheets of loosely-cohesive, large, and polygonal cells with distinct cell borders. The tumor cells demonstrate vesicular nuclei, single cherry-red nucleoli, and globular, eosinophilic, cytoplasmic inclusions. This tumor shows aggressive infiltration and extensive vascular invasion. The distinctive molecular characteristic of RTK is bi-allelic inactivation of the hSNF5/INI1 tumor-suppressor gene on the long arm or chromosome 22. Immunostaining for INI1 is uniformly lost in RTK, but is retained in most all other tumors that enter the differential diagnosis.
Primitive neuroectodermal tumor (PNET) of the kidney has been well documented. PNET can arise in patients ranging from 1 month to 72 years of age. Renal PNET is usually a large, poorly circumscribed grey-tan to white tumor that obliterates the uninvolved renal parenchyma. Hemorrhage and necrosis is common. Histologically, PNET consists of sheets of monotonous polygonal cells with high nuclear to cytoplasmic ratios. Mitotic figures are common. Tumor cells infiltrate adjacent renal parenchyma and pseudorosette formation can be seen. An immunohistochemical panel consisting of CD99, FLI-1, and WT-1 should be used to separate renal PNET from blastema-predominant nephroblastoma. Renal PNET usually demonstrates positivity for CD99 and FLI-1 and is negative for WT-1. Nephroblastoma usually expresses the opposite immunohistochemical findings. Molecular fusion studies demonstrate translocations of t(11;22), t(21;22) or others involving chromosome 22 in renal PNET.
Lymphoma and leukemia involving the kidneys should also be considered in the differential diagnosis; the most common setting is during the course of systemic lymphoma. This is known as secondary renal lymphoma. Primary renal lymphoma is rare but has been reported to account for 3% of surgically-excised renal tumors clinically thought to be renal carcinoma. Diffuse large B-cell lymphoma is the most commonly encountered renal lymphoma. Myeloid sarcoma refers to a neoplastic mass formed by proliferating myeloblasts at an extramedullary site, and rarely may be the primary occurrence of acute myeloid leukemia. When it involves the kidney it is usually a diffuse process that shows dense interstitial infiltration by immature granulocytes. An immunohistochemical panel including CD20, CD68, CD43, and MPO can help differentiate myeloid sarcoma from the other previously mentioned renal neoplasms.
Supplementary Questions:
- What immunohistochemical staining pattern is typical of metanephric adenoma?
- CK7–, CD57–, AMACR–, WT-1+v
- CK7–, CD57+, AMACR–, WT-1+
- CK7+, CD57–, AMACR+, WT-1–
- CK7+, CD57–, AMACR–, WT-1–
- What criterion is not used for unfavorable histology in nephroblastoma?
- Multipolar mitotic figures
- Nuclear enlargement
- Nuclear hyperchromasia
- Prominent nucleoli
- What molecular abnormality is characteristic of rhabdoid tumor?
- Deletion of 11p13 (WT-1 gene)
- Inactivation of hSNF5/INI1 gene
- Translocation of EWS/ERG gene
- Translocation of EWS/FLI-1 gene
References
- Bostwick DG, Cheng L, eds. Urologic Surgical Pathology. 3rd ed. Philadelphia, PA: Elsevier; 2014.
- Cheng L, Zhang DY, Eble JN, eds. Molecular Genetic Pathology. 2nd ed. New York, NY: Springer; 2013.
- Olgac S, Hutchinson B, Tickoo SK, et al. Alpha-methylacyl-CoA racemase as a marker in the differential diagnosis of metanephric adenoma. Mod Pathol. 2006;19:218-224.
Authors
2014
Stephanie Slemp, MD
Indiana University School of Medicine
Indianapolis, IN
Liang Cheng, MD FCAP
Surgical Pathology Committee
Indiana University School of Medicine
Indianapolis, IN
Answer Key
- CK7–, CD57+, AMACR–, WT-1+ (b).
- Prominent nucleoli (d).
- Inactivation of hSNF5/INI1 gene (b).