Clinical Summary
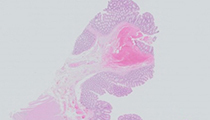
A 21-year-old male presents with intermittent rectal bleeding. A Hemoccult® test is positive. Colonoscopy demonstrates over 100 polyps throughout the colon. Further questioning reveals that both his father and grandfather died at an early age from colon cancer. Mutational testing for the APC gene is positive and a restorative proctocolectomy is performed.
Master List
- Familial adenomatous polyposis
- Juvenile polyposis syndrome
- Lynch syndrome
- MUTYH-associated polyposis
- Sporadic adenomas
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2014, case 12 and is familial adenomatous polyposis.
Criteria for Diagnosis and Comments
The slides show colonic mucosa with multiple areas of flat adenomatous change (polyps). There is no evidence of high grade dysplasia or carcinoma. These findings, in the context of the clinical history and molecular findings are typical for classic familial adenomatous polyposis (FAP).
FAP is an autosomal dominant genetic disorder that predisposes one to colorectal carcinoma. Those carrying this gene will with almost certainty develop colorectal carcinoma (approaching nearly 100%) if the colon is not removed. This disease is beset by numerous adenomas that usually develop in the second or third decades of life.
The term classic is used to differentiate the most common form from its variants. In classic FAP, at least 100 colorectal adenomas are present but this number may be much higher. The diagnosis of classic FAP can be made if any of the following criteria are met: 1) a germ-line disease causing mutation in the adenomatous polyposis coli (APC) gene; 2) 100 or more colorectal adenomas; 3) any number of colorectal adenomas at a young age in a person with a family history of FAP. Extracolonic gastrointestinal manifestations can include fundic gland polyps and adenomas of the duodenum/periampullary area.
Variants of FAP include attenuated FAP, Gardner syndrome and Turcot syndrome. In attenuated FAP the number of colorectal polyps is less than in the classic form with the average being around 30, but this number may vary widely. The other gastrointestinal findings of fundic gland polyps and duodenal/periampullary adenomas are similarly found. In the past, Gardner syndrome was used to describe colorectal polyposis with coexistent epidermoid cysts, osteomas and desmoids tumors. Similarly, Turcot syndrome was used to describe patients with colorectal polyposis and brain tumors (medulloblastoma, astrocytoma or glioblastomas). Interestingly, Turcot syndrome can be associated with either FAP or Lynch syndrome. These associations have a molecular basis with mutations of the APC gene or mismatch repair genes being found in individuals with this Turcot phenotype.
FAP accounts for less than 1% of all newly diagnosed colorectal carcinomas. APC germ-line mutations are the underlying etiology for FAP. This gene is ubiquitously expressed in all tissues and has complete penetrance; however the phenotype can vary within the same family. The APC gene is a tumor suppressor gene that is involved in multiple cellular functions including cell adhesion, signal transduction and activation. This gene is a negative regulator of the Wnt signaling pathway and is important in controlling B-catenin levels which is the main driver in FAP. Over 700 mutations have been identified to date with the most common occurring in the 5’ region. Most are frameshift or missense mutations (approximately 95%). FAP follows the classical two-hit model of carcinogenesis with 95% of those with FAP carrying germ-line mutations. Most carriers receive one aberrant APC allele and over time develop a second "hit" in another allele, within colonic epithelial cells. The relationship between phenotype-genotype is quite interesting with distinct associations present. However, mutations are not always found with only 60-80% of those with the classic phenotype found to have mutations; this is where the additional diagnostic criteria mentioned above come into play. Between 30 to 50% of all new FAP patients have a de novo underlying mutation of the APC gene or rarely exhibit APC mosaicism.
Colorectal adenomas and carcinomas in classical FAP can be found throughout the colon but do have a predisposition for the left side. Initially most patients are not symptomatic from adenomas, but over time as the size and number of adenomas increase rectal bleeding and diarrhea can occur as in our case. Colorectal adenomas are usually identified in the second or third decade. There is a greater than 90% chance that colorectal carcinoma will develop by the age of 50 in the untreated population with the mean age being 40. Grossly, colorectal adenomas are sessile, spherical and small (less than 5 mm in diameter). Large pedunculated adenomas are much less common. Adenomas found in FAP are histologically identical to that of their sporadic counterparts. Histologically, dysplastic aberrant crypt foci are the precursor to these adenomas and can be identified before adenomas are grossly seen. They are single crypts with dysplastic epithelial cells. If one or more dysplastic aberrant crypt foci are identified in a colon the diagnosis of FAP is extremely likely.
In FAP, adenomas can also occur throughout the small bowel, however the duodenum/periampullary area is the most common site involved. Adenomas at this site are felt to be the result of epithelial cell interaction with bile. These adenomas usually occur a decade later then colorectal adenomas. Fundic gland polyps of the stomach also occur. While fundic gland polyps in FAP do not inherently have adenomatous change, they are more likely to develop dysplasia then their sporadic counterparts with the incidence ranging from 9-50%.
The optimal care of individuals with FAP has some unique dilemmas. First is the screening of known carriers which starts around ages 10-15 years with sigmoidoscopy or colonoscopy. Once an adenoma is detected annual or biannual colonoscopy should be performed. The colon is usually removed in the late teens after secondary education for psychosocial reasons in asymptomatic carriers. The procedure performed varies between three depending on the desired outcome and form of FAP. Then procedures performed are restorative proctocolectomy; colectomy with ileorectal anastamosis; or colectomy with mucosal proctectomy, ileal pouch and ileo-anal pouch anastamosis. Even after colectomy carriers are not "out of the woods." Any residual rectum left behind can result in adenomas or eventually carcinoma. In addition there is a 4 to 10% risk of duodenal/periampullary adenocarcinoma.
The differential diagnosis includes any entities they have similar histological or clinical features. MUTYH-associated polyposis is similar to FAP in terms of clinical and histological manifestation. However, it is autosomal recessive in inheritance and caused by biallelic mutations in the MutY homologue gene (MUTYH) which is involved in base pair excision repair.
Sporadic adenomas are a diagnostic consideration but the clinical scenario and the number of adenomas would be highly unusual and the presence of the APC gene mutation excludes this process as well. Lynch Syndrome is another consideration in which a specific genotype-phenotype relationship occurs. These carriers usually have abnormalities in mismatch repair system involving MLH1, MSH2, PMS2, MSH6 or EPCAM. These carriers do have an adenoma/carcinoma sequence but the number of adenomas is not increased to the degree found in FAP. Juvenile polyposis is a consideration based on age but the histological features of cystically dilated branching glands with edematous stroma are not seen in FAP. Juvenile polyposis is due to underlying germ-line mutations in SMAD4 or BMPR1A in 50-60% of cases.
Supplementary Questions:
- Familial adenomatous polyposis can be caused by sporadic germline mutations.
- True
- False
- A 14-year-old girl with melena undergoes a colonoscopy. She is found to have seven colorectal polyps. Histologically these polyps are composed of cystically dilated branching glands, edematous stroma, and reactive epithelium. There is no adenomatous change. Further questioning reveals her 2 older siblings have also had melana and polyps. Which of the following genes is the most likely culprit in this scenario?
- APC
- EPCAM
- MSH2
- MUTYH
- SMAD4
- Which finding is not associated with familial adenomatous polyposis?
- Desmoid tumors
- Duodenal/ampullary adenocarcinoma
- Endometrial adenocarcinoma
- Glioblastoma
- Osteoma
References
- Bosman, FT, Carneiro F, Hruban RH, Theise ND. WHO Classification of Tumors of the Digestive System, Fourth Edition. Lyon, FR: Internal Agency for Research on Cancer Press; 2010.
- Fenoglio-Preiser CM, Noffsinger AE, Stemmermann. GN, Lantz PE, Isaacson PG, eds. Gastrointestinal Pathology: An Atlas and Text. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
- Montgomery EA, Voltaggio L. Biopsy Interpretation of the Gastrointestinal Tract Mucosa: Volume 2. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.
- Odze RD, Goldblum JR., Surgical Pathology of the GI Tract, Liver, Biliary Tract and Pancreas. Philadelphia, PA: Saunders Elsevier; 2009.
Author
2014
William V. Chopp, MD FCAP
Surgical Pathology Committee
Michigan Pathology Specialists
Grand Rapids, MI
Answer Key
- True (a).
- SMAD4 (e).
- Endometrial adenocarcinoma (c).