Clinical Summary
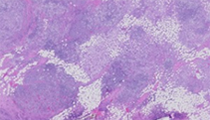
The patient is a 21-year-old woman with an approximately one-year complaint of progressive lymphadenopathy and malaise. She noticed a growing firm mass in her right breast, which is not fixed to the overlying skin. At resection, a 6.5 cm ill-defined firm pale mass is noted. Representative sections throughout the entire mass are submitted for histologic analysis.
Master List
- Diabetic mastopathy
- Extranodal Rosai-Dorfman disease
- Fibrous histiocytoma
- Idiopathic granulomatous mastitis
- Langerhans cell histiocytosis
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2015, case 05, and is extranodal Rosai-Dorfman disease.
Criteria for Diagnosis and Comments
Histologic sections consist of breast parenchyma diffusely infiltrated by a mixed inflammatory infiltrate. This consists of large histiocytes, lymphocytes, plasma cells, and scattered neutrophils. The infiltrate does not appear overtly lobulocentric. The histiocytic proportion is predominant, and consists of large cells with vesicular chromatin, indistinct nucleoli, and abundant pale to eosinophilic cytoplasm. Where the histiocytes are abundant, there is also fibrosis. Depending on the slide received, areas showing histiocytes with engulfed smaller inflammatory cells are prominent. Necrosis, significant cytologic atypia, or increased mitotic activity are not observed. These findings are morphologically compatible with extranodal Rosai-Dorfman disease (ERDD).
Rosai and Dorfman originally described a disease they termed 'sinus histiocytosis with massive lymphadenopathy'. Their first case series showed common clinical features; young African American women presenting with cervical lymphadenopathy, fever, leukocytosis, and hypergammaglobulinemia. The disease followed a protracted course but typically led to a complete recovery in most instances. In later years, this clinical picture broadened to include adult patients, other lymph node regions, as well as extranodal tumors (about one-third of cases). The most common clinical scenario still results in complete recovery. However, unusual cases of persistent lymphadenopathy and rarely death do occur. Death due to disease is usually associated with generalized lymphadenopathy and immune dysregulation. Aggressive chemotherapy has been tried with these cases, but an effective treatment remains elusive.
The etiology of ERDD is unknown. Given the typical clinical picture, an infectious etiology has long been postulated. Although viruses such as parvovirus B19 (especially intra-abdominal lesions), human herpesvirus-8 and Epstein-Barr virus have been implicated, no definitive causal etiology has yet been proven. Recently, similarities between ERDD and IgG4-related disease have been reported, such as increased numbers of IgG4+ plasma cells. However, other studies have failed to show a relationship.
When the disease presents in lymph nodes, the histologic findings are relatively constant; capsular fibrosis, sinusoidal dilatation with histiocytosis and mixed inflammatory cells, and effacement of follicular architecture. Perhaps the most striking histologic feature is the marked phagocytic activity of enlarged histiocytes with abundant clear cytoplasm. These cells can show engulfed lymphocytes, neutrophils, and even red blood cells. Most of the time the phagocytic activity consists of cells engulfed in intracytoplasmic vacuoles (a phenomenon termed emperipolesis), though nuclear debris can occasionally be seen. Significant cytologic atypia and necrosis are absent. Immunohistochemical studies reveal consistent S100 and CD68 positivity, as well as other monocytic markers. CD1a is typically negative. CD30 positivity has been occasionally reported. They are negative for cytokeratins and melanocytic markers other than S100.
ERDD more commonly involves the skin, paranasal sinuses, orbit, and soft tissue. Involvement of the breast is rare, with reports describing less than 10 cases in the literature. In the largest case series, most cases were localized to the breast. Bilateral tumors were reported, as well as some cases associated with generalized lymphadenopathy. Grossly, the tumors were typically circumscribed. Microscopically, there were variable amounts of fibrosis.
When the histiocytosis and emperipolesis is prominent, the diagnosis can be straight-forward. However, the histiocytosis can aggregate and resemble dense fibrosis (as in this case), and obscure the typical emperipolesis. When this happens, other diagnoses may enter the differential. Fibrous histiocytoma can be found on the trunk or extremities. It typically shows a proliferation of elongated spindle cells arranged in a storiform pattern, with variable numbers of giant and foam cells. Some cases can show a prominent foam cell component that may bear a passing resemblance to the histiocytes of ERDD. However, the lesional cell is S100 negative, which will readily distinguish it from ERDD.
Diabetic mastopathy can form discreet masses or diffuse nodularity. It usually occurs in premenopausal women with a long-standing history of type I diabetes. The typical histologic findings include a myofibroblastic proliferation and predominantly mononuclear inflammation. When numerous, the myofibroblasts can appear enlarged and epithelioid, which may resemble aggregates of histiocytes in ERDD. However, the inflammation in diabetic mastopathy is lobulocentric and consists mainly of small lymphocytes, which are features not observed in ERDD.
Idiopathic granulomatous mastitis can also cause palpable breast masses. Occurring in young parous women, it also consists of mixed inflammatory cells with variable amounts of fibrosis. However, inflammation consists of lobulocentric granulomas with neutrophils and focal microabscess formation. A histiocytosis resembling that of ERDD is not observed.
Langerhans cell histiocytosis is a histiocytic proliferation derived from Langerhans cells. When occurring in the lymph node, the low power morphology resembles that of ERDD. It also occurs in extranodal sites, where it shows aggregates of histiocytes and mixed inflammatory cells, especially eosinophils. The characteristic morphologic finding is cells with irregular elongated nuclei with prominent grooves and abundant pale/eosinophilic cytoplasm. Like ERDD the histiocytes are positive for S100, but they are also positive for CD1a. CD1a positivity in ERDD has rarely been reported. However, in cases where ERDD and Langerhans cell histiocytosis occur together, CD1a is negative in ERDD. Also, emperipolesis is not observed.
Supplementary Questions:
- Which of the following is most correct concerning Rosai-Dorfman disease?
- Current consensus opinion is that it should be classified as part of IgG4-related disease.
- It is relatively common in the breast.
- Morbidity and death are rare, but when they occur they are typically associated with immune dysregulation.
- Most cases appear to be associated with a viral etiology.
- Which of the following is least correct concerning the pathologic findings of Rosai-Dorfman disease?
- CD30 positivity can occasionally be seen.
- Cytologic atypia and necrosis are common.
- Emperipolesis of granulocytes and red blood cells can occur.
- Histiocytosis and emperipolesis are nearly constant findings in both nodal and extranodal Rosai-Dorfman disease.
- Which of the following best differentiates Rosai-Dorfman disease and Langerhans cell histiocytosis?
- CD1a negativity
- S100 positivity
- Extranodal location
- Mixed inflammatory infiltrate
References
- Al-Daraji W, Anandan A, Klassen-Fischer M, et al. Soft tissue Rosai-Dorfman disease: 29 new lesions in 18 patients, with detection of polyomavirus antigen in 3 abdominal cases. Ann Diagn Pathol. 2010;14(5):309-16.
- Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. Semin Diagn Pathol. 1990;7:19-73.
- Green I, Dorfman RF, Rosai J. Breast involvement by extranodal Rosai-Dorfman disease: report of seven cases. Am J Surg Pathol. 1997;21(6):664-668.
- Ioachim HI, Medeiros JI. Ioachim's Lymph Node Pathology. 4th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2009.
- Liu L, Perry AM, Cao W, et al. Relationship between Rosai-Dorfman disease and IgG4-related disease: study of 32 cases. Am J Clin Pathol. 2013;140(3):395-402.
- Morkowski JJ, Nguyen CV, Lin P, et al. Rosai-Dorfman disease confined to the breast. Ann Diagn Pathol. 2010;14(2):81-87.
- O'Malley D, Duong A, Barry T, et al. Co-occurrence of Langerhans cell histiocytosis and Rosai-Dorfman disease: possible relationship of two histiocytic disorders in rare cases. Mod Pathol. 2010;23(12):161623.
- Rosai J and Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: A pseudolymphomatous benign disorder. Cancer. 1972;30:1174-1188.
- Schnitt SJ, Collins LC. Biopsy Interpretation of the Breast. Philadelphia, PA: Lippincott Williams and Wilkins; 2009.
- Swerdlow SH, Campo E, Harris NL, et al (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: IARC Press; 2008.
Author
2015
Brad B. Bryan, MD
Surgical Pathology Committee
Bend, OR
Answer Key
- Morbidity and death are rare, but when they occur they are typically associated with immune dysregulation (c).
- Cytologic atypia and necrosis are common (b).
- CD1a negativity (a).