- Home
- Member Resources
- Pathology Case Challenge
- Adrenal Gland
Clinical Summary
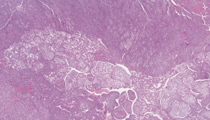
A 49-year-old woman presents with abdominal pain. Workup reveals a left adrenal mass and surgical intervention is undertaken. The resection specimen demonstrates a 15.0 cm mass replacing the adrenal gland, with local infiltration into soft tissue, kidney, and spleen. Several large-caliber veins grossly contain tumor thrombi. Cut section shows extensive necrosis and hemorrhage.
Master List
- Adrenal cortical adenoma
- Adrenal cortical carcinoma
- Metastatic carcinoma
- Metastatic melanoma
- Pheochromocytoma
- Renal cell carcinoma
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2013, case 28, and is an adrenal cortical carcinoma.
Criteria for Diagnosis and Comments
This is a poorly differentiated neoplasm with varied architectural patterns ranging from broad trabecular cords with slit-like sinusoids to solid and nested areas. Tumor cells are pleomorphic with distinct cell borders and varied cytoplasm, ranging from pale, finely vacuolated to densely compact and eosinophilic. Nuclei are eccentric, with significant pleomorphism, irregular chromatin and large prominent nucleoli. Mitotic activity is evident. Lymphovascular space invasion is present, including intraluminal involvement of medium size veins. Extensive necrosis is noted. These findings are characteristic of adrenal cortical carcinoma (ACC).
ACCs are rare tumors, with 1-2 cases per million population and with no gender predilection. Although primarily a tumor found in adults, occasional pediatric cases occur. Patients classically present with abdominal pain, discomfort, fullness or weight loss. Occasionally, ACC may secrete active hormones, and present with symptoms of virilization or hypertension. Imaging studies typically will show a heterogeneous mass, frequently with necrosis or calcifications. Tumors tend to be large and bulky, averaging 12.0-16.0 cm and can weigh over 1200.0 g. Invasion of major veins and adjacent structures is common. On cut surface, the tumor surface may have a variegated appearance, with hemorrhage, cystic degeneration, and necrosis.
Microscopically, ACC can display a variety of growth patterns, which makes the diagnosis challenging, particularly on core biopsy. As in this case, architectural patterns can include broad anastomosing trabecular cords with slit-like sinusoids, perithelial arrangement, long slender cords, nested, pseudopapillary, alveolar, or diffuse and solid. The stroma may show myxoid changes, fibrosis or calcification. Necrosis and invasion into the veins, sinusoids, and capsule are commonly seen. Cytologic features include nuclear pleomorphism and hyperchromasia. Nuclei may be hyperlobated or multinucleated with prominent multiple nucleoli. Nuclear pseudoinclusions can be seen. Brisk mitoses (greater than 5 per 50 high power fields), particularly as atypical forms, are characteristic. The cytoplasm may be lipid-rich imparting a pale vacuolated appearance or more compact and eosinophilic. Intracytoplasmic lipofuscin and hyaline globules can also be present.
Panels of immunohistochemical studies can be used to confirm the diagnostic impression of ACC. Tumor cells can be positive for Melan A, inhibin, calretinin, synaptophysin, vimentin and steroidogenic factor-1 (SF-1), while are usually negative for cytokeratin, EMA and chromogranin.
The differential diagnosis of ACC includes adrenal cortical adenoma, renal cell carcinoma, pheochromocytoma, and metastatic tumors, most commonly carcinomas and melanoma. Adrenal cortical adenomas are often smaller in size, and lack the features typical of ACC including nuclear pleomorphism, high mitotic rate, local invasiveness and metastatic spread. As the distinction between the two can sometimes be challenging, the Weiss system was developed to aid in the classification of adrenal cortical neoplasms. Nine histopathologic characteristics were identified as potentially useful in this setting which include: high nuclear grade, mitotic rate greater than 5 per 50 high power fields, atypical mitoses, clear cells comprising < 25% of tumor, diffuse architecture > 1/3 of tumor, necrosis, invasion of venous structures, invasion of sinusoidal structures, and invasion of capsule of tumor. The presence of three or more of these features correlates with malignant behavior. The three most specific features of malignancy are mitotic rate greater than 5 per 50 high power fields, atypical mitoses, and invasion of venous structures. High proliferation index by Ki67 favors a diagnosis of carcinoma, although a specific cutoff value has yet to be established. Of note, the above criteria do not apply to pediatric adrenocortical tumors where neoplasms with otherwise "aggressive" morphologic features can behave in an indolent manner.
Renal cell carcinoma can locally invade into and metastasize to the adrenal gland, and if high grade, show overlapping morphology with ACC. Clinical information, including imaging studies, can be useful in separating the tumors. If necessary, immunohistochemical panels can be used, as renal cell carcinomas are generally negative for the typical ACC markers, while positive for such markers as cytokeratin, EMA, RCC and CD10. As many types of malignant tumors can spread to the adrenal gland, one should always consider a metastatic carcinoma or melanoma. Clinical history can be very important in this situation. Immunohistochemical panels are useful in these differentials; utilizing an ACC profile in combination with keratin, EMA and primary site specific markers (i.e. lung, breast, enteric, etc) should readily define the primary. Although ACC can express the melanoma marker Melan A, true metastatic melanoma should not express other adrenocortical markers such as calretinin, synaptophysin, or SF-1.
Pheochromocytoma can also enter into the differential of ACC, as there can be overlap of gross, microscopic and cytologic features in high grade tumors. Immunohistochemical studies can also assist in separating the two, as pheochromocytomas are positive for chromogranin and negative for inhibin, calretinin and SF-1, which is opposite to that of ACC.
Supplementary Questions
- A 41-year-old woman presents with an 11.0 cm adrenal mass. The most reliable indicator of malignancy is?
- 20 mitoses per 50 high power fields with atypical mitoses
- Capsular invasion
- Marked nuclear pleomorphism and anaplasia
- Presence of metastasis
- Presence of necrosis
- Which of the following statements is true concerning adrenal cortical carcinoma?
- Capsular and vascular invasion are commonly found in benign tumors
- Patients frequently present with Cushing syndrome and/or virilization due to the large size of the tumor
- Pediatric tumors with necrosis, high mitotic rate with atypical mitoses, and size greater than 100 grams generally have better prognosis than adult tumors with similar features
- This tumor shows a female preponderance of 2:1
- Tumors weighing greater than 100 g are invariably malignant
- Adrenal cortical tumors are typically positive for all of the following except?
- EMA
- Inhibin
- Steroidogenic factor-1
- Synaptophysin
- Vimentin
References
- Aubert S et al. Weiss System Revisited: A Clinicopathologic and Immunohistochemical Study of 49 Adrenocortical Tumors. Am J Surg Pathol. 2002 26(12):1612-1619.
- Duregon et al. Diagnostic and Prognostic Role of Steroidogenic Factor 1 in Adrenocortical Carcinoma: A Validation Study Focusing on Clinical and Pathologic Correlates. Hum Pathol. 2012.
- Enriquez ML et al. The Use of Immunohistochemical Expression of SF-1 and EMA in Distinguishing Adrenocortical Tumors from Renal Neoplasms. Appl Immunohistochem Mol Morphol. 2012; 20(2):141-145.
- Lack, E. Tumors of the Adrenal Glands and Extraadrenal Paraganglia: AFIP Atlas of Tumor Pathology ARP Press, 2007.
- Lau S, Weiss L. The Weiss System for Evaluating Adrenocortical Neoplasms: 25 Years Later. Hum Pathol. 2009;40:757-768.
- Lloyd RV Adrenal Cortical tumors, Pheochromocytomas and Paragangliomas. Mod Pathol. 201124:S58-65.
- Sangoi, AR, McKenney, JK. A Tissue Microarray-Based Comparative Analysis of Novel and Traditional Immunohistochemical Markers in the Distinction Between Adrenal Cortical Lesions and Pheochromocytoma. Am J Surg Pathol. 2010;34:423-432.
- Weiss, LM. Comparative Histologic Study of 43 Metastasizing and Non-Metastasizing Adrenal Cortical Tumors. Am J Surg Pathol. 1984;8:163-169.
Authors
2013
Elena Chang, MD
Genitourinary Pathology Fellow
Cedars-Sinai Medical Center
Los Angeles, CA
Daniel J. Luthringer, MD
Surgical Pathology Committee
Cedars-Sinai Medical Center
Los Angeles, CA
Answer Key
- Presence of metastasis (d).
- Pediatric tumors with necrosis, high mitotic rate with atypical mitoses, and size greater than 100 grams generally have better prognosis than adult tumors with similar features (c).
- EMA (a).