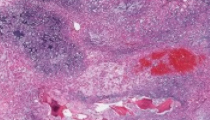
A 57-year-old man with no significant past medical history presents to a neurosurgeon for intractable lower back pain. Radiographic studies document a lytic mass within the sacrum compressing surrounding nerves. An image guided needle biopsy is performed and surgical excision is subsequently scheduled. A 5.8 centimeter soft, multilobulated variegated gray to gray-tan mass with a gelatinous texture was received for gross examination. Samples are prepared for routine histologic examination and electron microscopy. Tumor cells show staining for periodic acid Schiff staining which digests upon diastase treatment and are immunoreactive for S-100, pan-cytokeratin, EMA, and brachyury.
Master List
- Chondrosarcoma
- Chordoma
- Myxopapillary ependymoma
- Renal cell carcinoma
- Signet ring cell carcinoma
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2013, case 03, and is a chordoma.
Criteria for Diagnosis and Comments
Chordoma is the diagnosis in this case. Chordoma represents a rare tumor derived from the remnant notochord and represents the fourth most common malignant bone tumor following osteosarcoma, chrondrosarcoma, and Ewing sarcoma. This tumor affects males and females equally and primarily occurs in the fifth and sixth decades. Chordoma primarily affects the sacrococcygeal and sphenooccipital regions, but they may also occur in the cervical and lumbar regions. Patients affected by this tumor usually present with pain and neurologic deficits related to the anatomic site affected. The primary treatment modality is wide en bloc excision of the tumor, but tumors that cannot be surgically excised are typically treated with adjuvant radiation therapy. Grossly, chordomas have a glistening blue-gray cut surface separated by fibrous septae. The characteristic histologic findings in chordomas are large polygonal cells with distinct borders wrapping around themselves and the vacuolated physaliphorous cell, the term deriving from the Greek physalis, or “bubble.” Tumor cells grow in small nests and cords within a myxoid matrix and demonstrate round uniform nuclei with low nuclear-to-cytoplasmic ratios. Chordomas typically demonstrate immunohistochemical staining for pan-cytokeratin, S-100 protein, epithelial membrane antigen (EMA), HBME-1, brachyury, SOX9, SHH (sonic hedgehog), cathepsin K, and cadherin. Tumor cells are typically negative for cytokeratin-7 and cytokeratin-20. Chordomas are associated with a hypodiploid karyotype with frequent loss of chromosome 3p, 4, 10 and 13. Comparative genomic hybridization studies have documented loss of chromosomes 1p, 3p, and chromosomal gains in 5q, 7q, 12q, and 20. The candidate chromosomal region associated with both inherited (rare) and sporadic forms of chordoma has mapped to chromosome 1p36.13.
There are no known associations of chordoma with inherited syndromes. Chordomas are associated with tumor recurrence and a high mortality rate. The mean patient survival is 4 years. Metastatic disease occurs late in the disease course, with lungs and skin being the most common sites for disease spread. The primary differential diagnostic considerations, especially in the context of a destructive bone tumor of the sacrum, include chrondrosarcoma, myxopapillary ependymoma, metastatic clear cell carcinomas, and giant cell tumors.
Chondrosarcoma, malignant tumors of hyaline cartilage, may also occur in the sacrum (where they present as eccentric rather than midline lesions) and are associated with bony destruction, but these tumors more commonly occur in the pelvis and proximal extremities (e.g. humerus and femur). Like chordoma, chondrosarcoma also typically affects older adults, especially between the 5th and 7th decades of life. The microscopic discrimination of chondrosarcoma, particularly the myxoid variant, and chordoma may be particularly challenging in the context of small biopsy specimens where inadequate sampling becomes a major diagnostic limitation. Microscopically, the myxoid matrix, cytologic features, and lobular growth pattern present in chondrosarcoma may be confused with similar features of chrodoma; however, the distinctive physaliphorous cells of chordoma are not identified in chondrosarcomas. Immunohistochemical studies can be quite helpful in distinguishing between chrondrosarcoma and chordoma. Chondrosarcomas are typically immunoreactive for D2-40 (podoplanin) and EMA, but negative for pan-cytokeratin and glial fibrillary acidic protein (GFAP). In contrast, chordoma is typically positive for EMA and pan-cytokeratin,and brachyury, but negative for D2-40 and GFAP.
Metastatic carcinomas to the sacrum such as clear cell renal cell carcinoma and mucinous adenocarcinomas, especially signet ring carcinoma of rectum, may also enter into the differential diagnosis with chordoma, but metastatic tumor deposits are oftentimes smaller and multiple, in contrast to the larger mass typically associated with chordoma. The vacuolated and/or optically clear cytoplasmic features identified in metastatic carcinomas may be confused with the abundant and vacuolated appearance of the physaliphorous cells of chordoma. In this differential, immunohistochemical staining for markers such as pan-cytokeratin and EMA would demonstrate staining in both carcinoma and chordoma, but these two entities could be discriminated by the presence of staining for S-100 protein and brachyury in chordoma. In addition, clear cell renal cell carcinomas are typically immunoreactive for Pax-2, Pax-8, and CD10, while metastatic mucinous adenocarcinomas such as signet ring cell carcinoma of the rectum would show prominent mucicarmine staining and a cytokeratin-7 negative, cytokeratin-20 positive profile with nuclear CDX-2 staining.
Myxopapillary ependymoma, considered a grade I glial tumor by the World Health Organization, is also a diagnostic consideration given the location of its occurrence in the sacrococcygeal region and its similar histologic features to chordoma. Myxopapillary ependymoma is almost exclusively restricted to the conus medullaris and filum terminale and may present with clinical signs similar to chordoma including pain and neurologic deficits resulting from nerve compression. This tumor is characterized by hyalinized stromal blood vessels surrounded by mucinous material and microcysts separating tumor cells from the vascular core. While the histologic features of myxopapillary ependymoma are classic for its diagnosis, prominently myxomatous variants of the tumor may be mistaken for chordoma, particularly in small biopsy samples. Unlike the natural history of chordoma, myxopapillary ependymomas are indolent and surgical cure is possible. Tumor cells of myxopapillary ependymoma are typically immunoreactive for S-100 protein and GFAP, as well as pan-cytokeratin. While chordomas may also demonstrate staining for pan-cytokeratin and S-100 protein, no staining for GFAP is present.
Supplementary Questions:
- Chordomas are considered tumors of notochordal vestiges and most commonly affect patients in the 2nd and 3rd decades of life.
- True
- False
- Chordoma can typically be distinguished from chondrosarcoma by which of the following immunohistochemical stains:
- Brachyury, D2-40, and pan-cytokeratin
- Galectin-3, pan-cytokeratin, and S-100
- GFAP, pan-cytokeratin, and CD99
- S-100, chromogranin, and cytokeratin-7
- S-100, pan-cytokeratin, and GFAP
- Which of the following statements concerning chordoma is false?
- Chordomas classically demonstrate physaliphorous cells on histologic examination
- Chordomas typically affect younger adults and have no risk for recurrence
- Chordomas may be distinguished from myxopapillary ependymoma by immunohistochemical staining characteristics
- The candidate gene locus for chordoma has been mapped to chromosome 1p
- The primary treatment approach for chordoma is en bloc excision with negative margins
References
- Bone and Joints In: Rosai J, ed. Rosai and Ackerman's Surgical Pathology. Philadelphia, PA: Elsevier;2011:2055-2057.
- Cho HY, Lee M, et al. Immunohistochemical comparison of chordoma with chondrosarcoma, myxopapillary ependymoma, and chordoid meningioma. Appl Immunohistochem Mol Morphol 2009;17(2):131-138.
- Horvai A. Notochordal tumors In: Folpe A and Inwards CY, eds. Bone and Soft Tissue Pathology. Philadelphia: Saunders Elsevier;2010:408-413.
- Takei H and Powell SZ. Novel immunohistochemical markers in the diagnosis of nonglial tumors of nervous system. Adv Anat Pathol 2010;17(2):150-153.
Author
2012
Noel Brownlee, MD, PhD FCAP
Surgical Pathology Committee
Bon Secours Saint Francis Health System
Greenville, SC
Answer Key
- False (b).
- Brachyury, D2-40, and pan-cytokeratin (a).
- Chordomas typically affect younger adults and have no risk for recurrence (b).